•Infantile fibrosarcoma (IFS) is histologically identical to
classic fibrosarcoma of adults, but carries a much more
favourable prognosis.
• It occurs in infants and young children, metastasizes
rarely, and has similar history of fibromatoses.
•IFS is morphologically and genetically related to congenital
mesoblastic nephroma.
Epidemiology:
•IFS accounts for 36%-80% of congenital cases and
36%-100% occurs in first year of life.
•IFS is rare after 2 years of age.
•There is slight male predominance.
Sites of involvement:
• The superficial and deep soft tissue of the extremities,
especially distal (61%), followed by the trunk (19%) and
head and neck (16%).
Clinical findings:
•IFS presents as a solitary enlarging, nontender mass or
swelling in the soft tissues and grows rapidly.
•The diameter may exceed 30 cm.
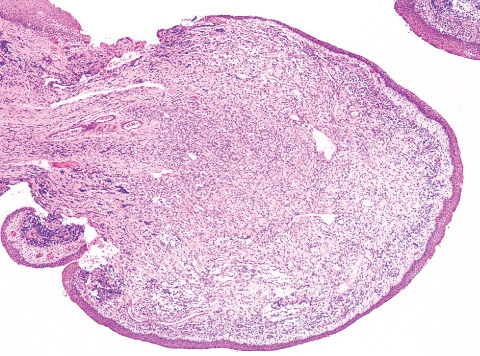
Gross:
•poorly circumscribed, lobulated mass infiltrating adjacent
soft tissue.
•The cut surface is soft to firm, fleshy, and grey to tan with
variable areas of myxoid or mucinous change, cystic
degeneration, haemorrhage, necrosis, and yellow-red.
discoloration.
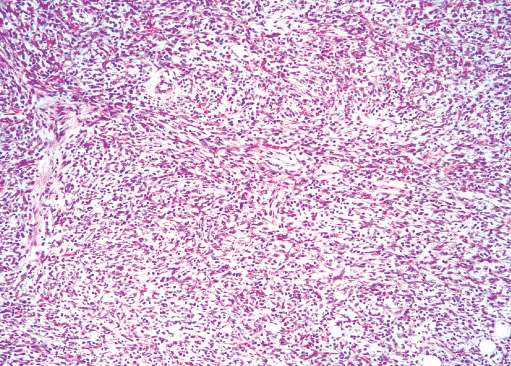
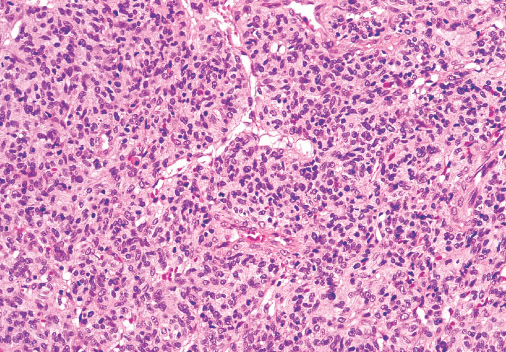
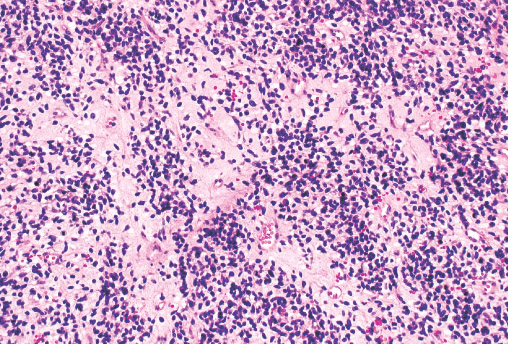
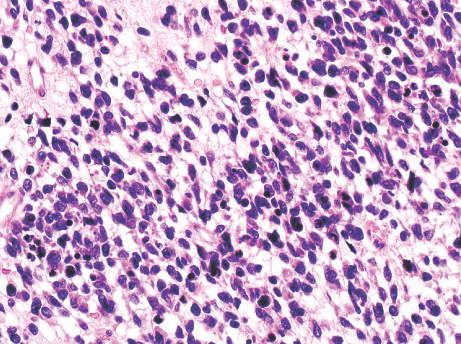
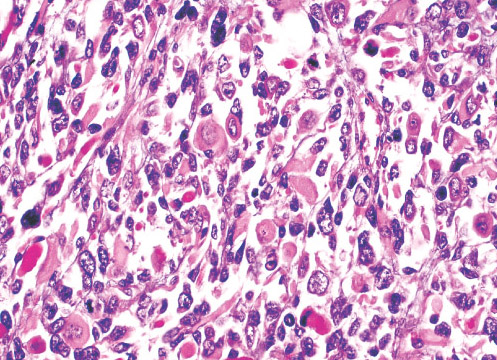
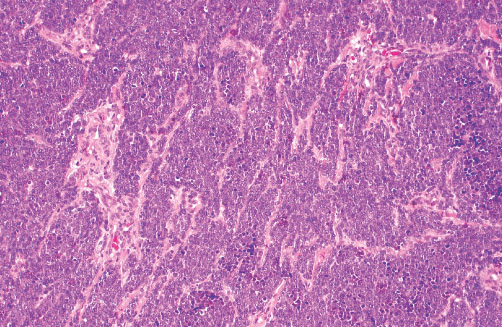
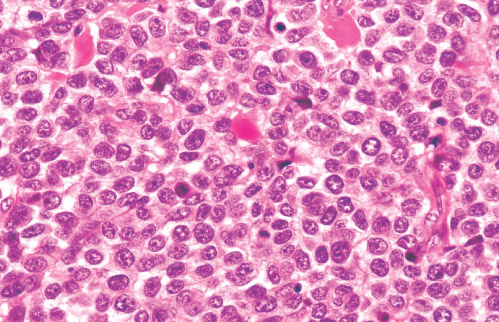
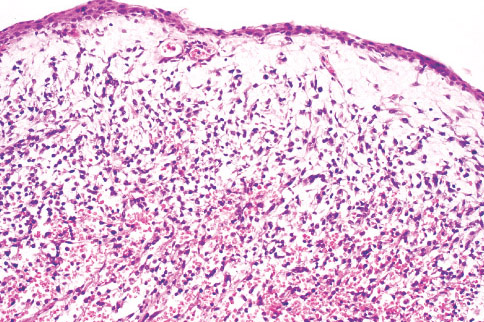
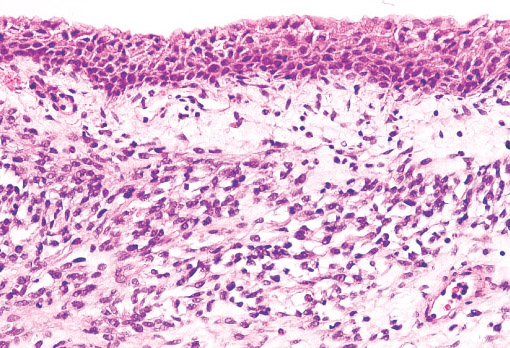
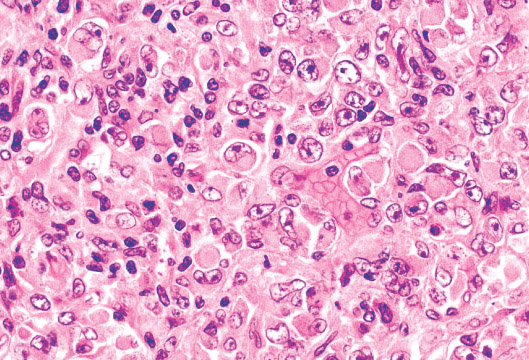
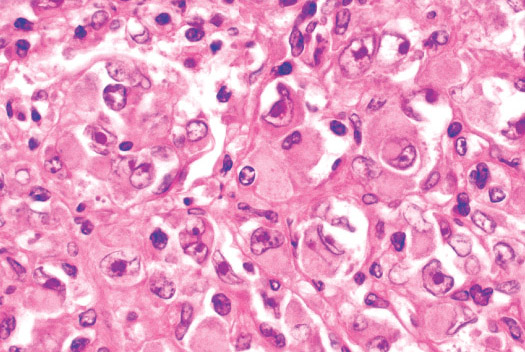
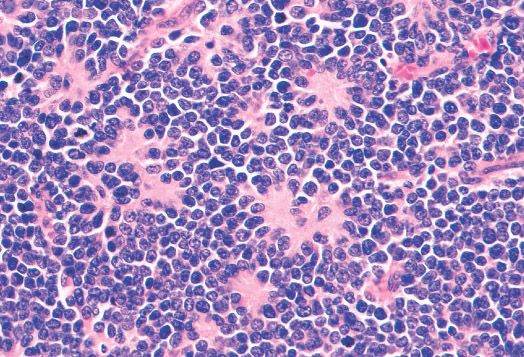
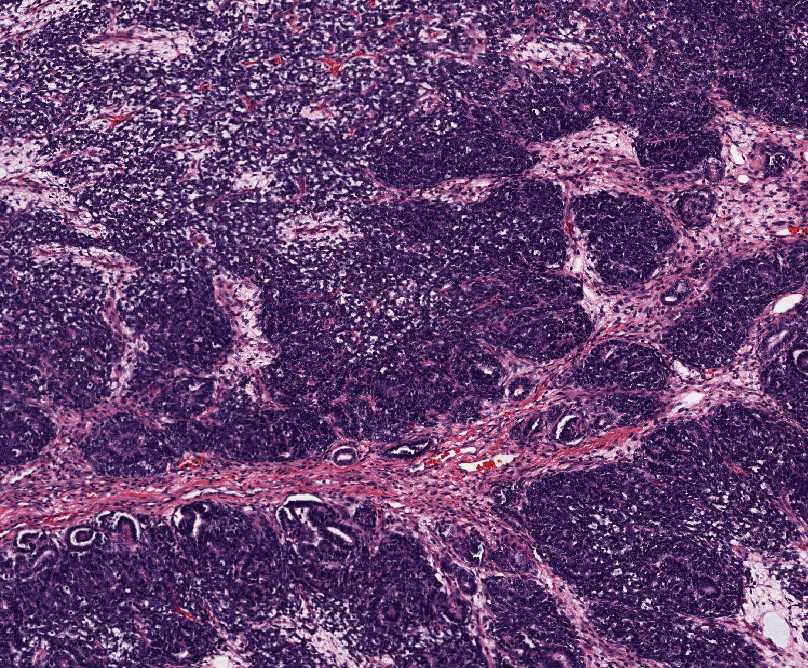
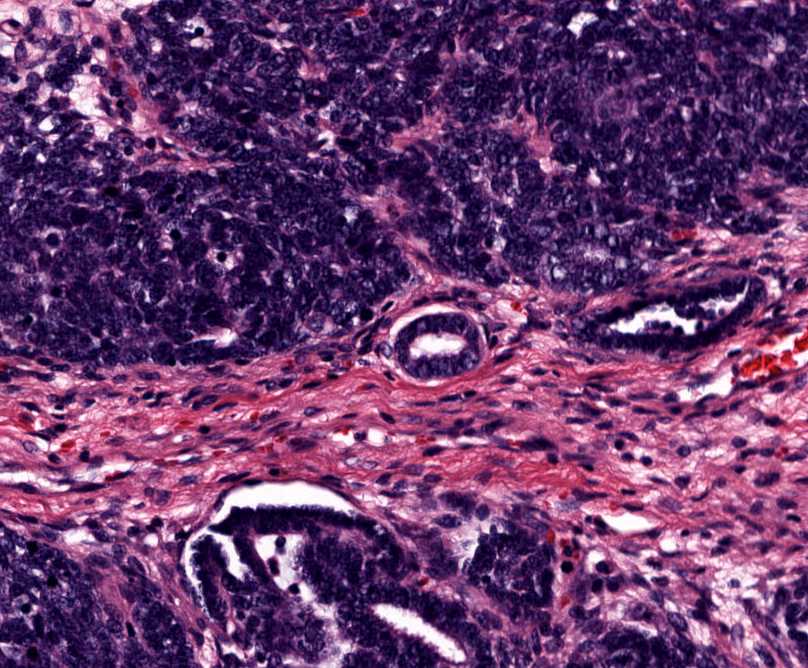
Histopathology:
•IFS show dense cellularity composed of intersecting
fascicles of primitive ovoid and spindle cells with
herringbone pattern or forming interlacing cords, sinous
bands or sheets of cells.
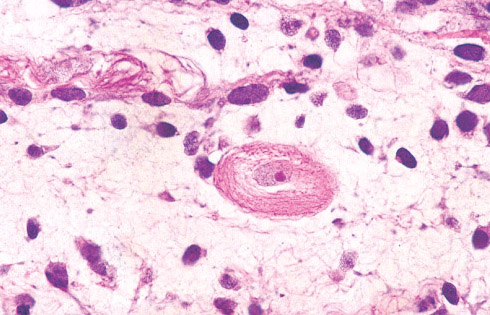
•Zonal necrosis or hemorrhage are frequent and may be
associated with dystrophic calcifications.
•Cells show little pleomorphism.
•Collagen production is variable, and mitotic activity is
prominent.
• Most IFS show scattered chronic inflammatory cells and
may display focal extramedullar haematopoiesis.
•Histological variations may show areas of
haemangipericytoma-like pattern, myxoid changes or
predominant immature ovoid cells proliferation with minimal
collagen.
• Infiltrative growth pattern may result in entrapmet of
adipose and skeletal tissue.
Immunophenotype:
•Positive for vimentin (100%), neuron-specific enolase NSE
(35%), smooth muscle actin (33%).
Genetics:
•Translocation t(12;15).
Prognosis:
• IFS has a more favourable outcome compared with adult
fibrosarcoma.
•The mortality ranges from 4% to 25%, and the recurrence
rate is 5% to 50%.
The information provided on this site is designed to support, not replace, the relationship that exists between a patient/site visitor and his/her existing physician. Banners on the Home Page and at the top of other pages are advertisements. Our editorial content is free of any commercial influence. There are no medical or personal patient informations included. Disclaimer: this information is intended for pathologists, orthopedic surgeons and laboratory personnel, who understand that medical information is often imperfect, and must also be interpreted in the context of a patient's clinical data using reasonable medical judgment.
The site is supported by advertisement. I value our advertisers, who make this free website possible.
Pedorthpath.com is concerned with online privacy for all visitors and advertisers at our site. Any information sent to us by email or otherwise, except for information intended to be posted at the website, is treated as confidential, and will not be disclosed to any third party without the express permission of the visitor or advertiser.
Last modification date: 03/15/2010.
Site designed and maintained by Dariusz Borys, M.D
RHABDOMYOSARCOMA
EMBRYONAL RHABDOMYOSARCOMA
Definition:
•A primitive, malignant soft tissue sarcoma with phenotypic
and biological features of embryonic skeletal muscle.
Epidemiology:
•Most common subtype of rhabdomyosarcoma, occuring in
3.0/milion U.S. children <15 years old.
•Most (46%) of embryonal rhabdomyosarcomas occurs in
children less than five year old.
• Male to female ratio is 1.2:1.0.
Sites of involvement:
•Most common location head and neck (47%) followed by
genitourinary system (28%).
•Common location in the genitourinary tract include the
urinary bladder, prostate and paratesticular soft tissues.
•Typical sites involving head and neck area include soft
tissue surrounding the orbit and eyelid, oropharynx,
parotid, auditory canal and middle ear, pterygoid fossa,
nasopharynx, nasal passages and paranasal sinuses,
togue and cheek.
Clinical findings:
•Clinical symptoms are associated with the affected area.
•Head and neck lesions can cause proptosis, diplopia,
sinusitis or unilateral deafness, depending on their location.
•Similarily, genitourinary lesions may produce scrotal mass
or urinary retention, and biliary tumors may cause
jaundice.
Gross:
•Like most primitive pediatric tumors, embryonal
rhabdomyosarcomas form poorly circumscribed, fleshy,
pale tan masses that directly invade adjacent soft tissue.
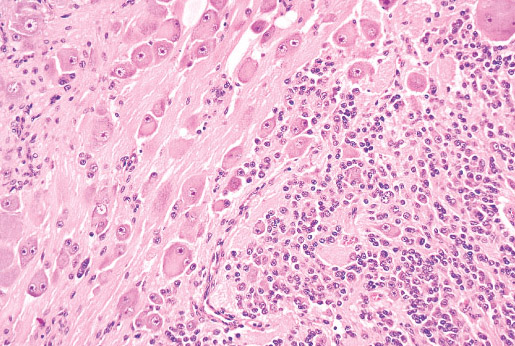
Histopathology:
•Embryonal rhabdomyosarcomas are composed of primitive
mesenchymal cells in various stages of myogenesis
(rhabdomyoblasts).
•Stellate cells with lightly amphophilic cytoplasm and
central, oval nuclei represent the most primitive end of this
spectrum.
• As these cells differentiate they progressively become
more elongated with eosinophilic cytoplasm also called
such as "tadpole", "strap", and "spider" cells.
• Bright eosinophilia, cytoplasmic cross striations, and
multinucleation indicate terminal differentiation, and
myotube forms may be present.
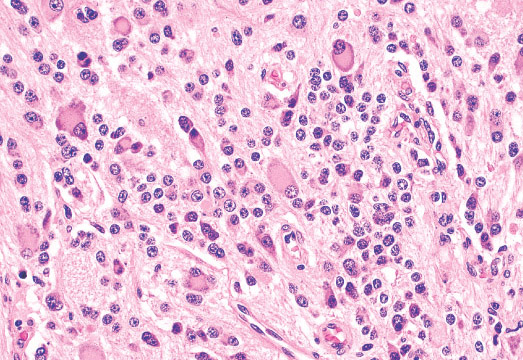
• Densely arrayed whorls of fascicles of spindle cells
constitute the spindle cell variant of embryonal
rhabdomyosarcoma.
• These spindle cells often resemble smooth muscle cells,
with blunted central nuclei and tapered ends, but
cytoplasmic cross striations, if present, and/or broght
pattern and mixed alveolar/embryonal. All alveolar
rhabdomyosarcomas exhibit round cell cytological features
resembling lymphoma, but with primitive myoblastic
differentiation.
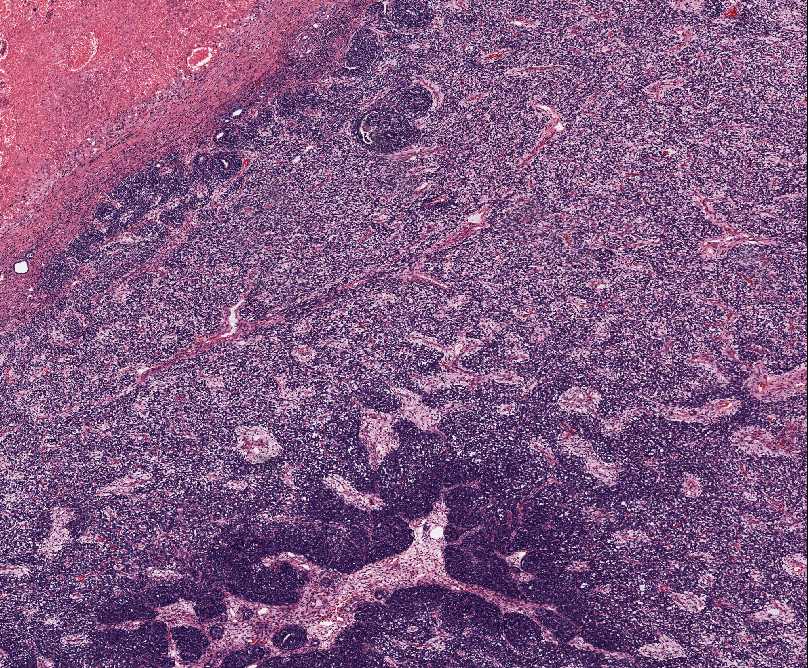
1)Typical alveolar rhabdomyosarcomas produce fibrovascular septa that separate the tumor cells into discrete nests. These cells contain central clusters of cells with loss of cohesion at the periphery. Tumor cells align the septa in the picket fence pattern. Giant cells with rhadbomyoblastic differentiation are common.
2)Solid variant of ARS lack the fibrovascular stroma and form sheets of round cells with variable rhabdomyoblastic differentiation. Occasionally small nests may be seen with larger samples.
3)Mixed embronal/alveolar rhabdomyosarcoma contain foci with embryonal histology (mixed stroma with spindle cell myoblasts), as well as areas of alveolar histology.
Immunophenotype:
• tumor cells are positive for desmin, myogenin, myo-D.
Genetics:
•Most common translocation t(2;13)(q35;q14) PAX3/FKHR,
and less common t(1;13)(p36;q14) PAX7/FKHR.
Prognosis:
•Alveolar rhabdomyosarcomas are high grade sarcomas and
are more aggressive than embryonal rhabdomyosarcoma.
•Usually presents as large abdominal mass felt by mother
holding child.
Gross:
• Large, solitary, well-circumscribed mass (10% bilateral or
multicentric), soft, homogenous, tan-gray.
• May show hemorrhage, necrosis, cysts and lobular pattern.
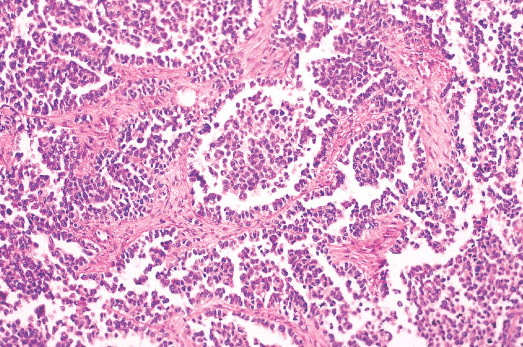
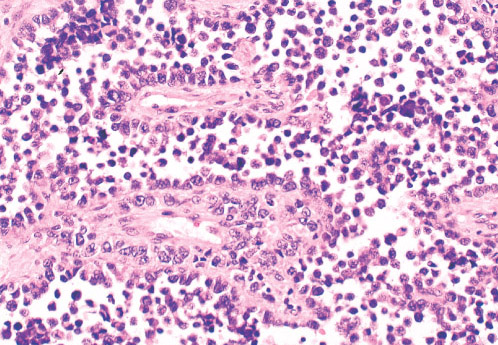
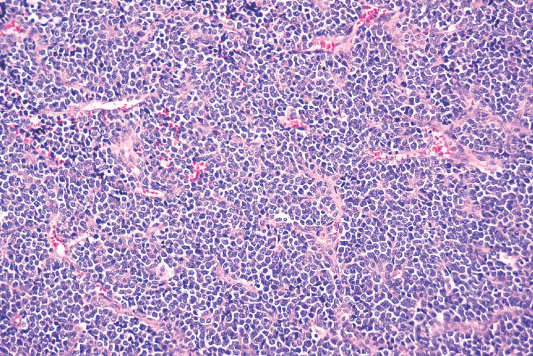
Histopathology:
• Triphasic with undifferentiated blastema (cellular with small
blue primitive cells with scanty cytoplasm, nuclei are
overlapping with finely dispersed chromatin; patterns are
diffuse, nodular, cordlike or basaloid), fibroblast-like stroma
and epithelium (abortive tubules, glomeruli with
elongate/ovoid nuclei having molded/wedged shapes).
• May show additional findings of smooth muscle, cartilage,
adipose tissue, squamous or mucinous epithelium, bone,
neural tissue.
• Anaplastic cells may be found in 5% of cases and are
associated with worse prognosis.
Immunohistochemistry:
• Positive for WT1, desmin and focally vimentin.
Genetics:
•Abnormal expression of WT1 (11p13).
Staging (National Wilms Tumor Study Group)
•Stage I (43%): tumor limited to kidney and completely resected, renal capsule intact, tumor not ruptured or biopsied prior to removal, no residual tumor beyond margins of resection, no tumor within renal vein (tumor within intrarenal vessels is OK), no nodal involvement or distant metastases
•Stage II (23%): tumor extends beyond kidney but is completely resected, regional extension of tumor (vascular invasion outside of renal parenchyma or within the renal sinus, or capsular penetration but with negative surgical margin), operative tumor spill confined to flank (no peritoneal contamination), tumor biopsy (except FNA) prior to surgery
•Stage III (23%): nonhematogenous metastases to abdomen only (such as regional lymph nodes), including tumor implants in or penetrating peritoneum; gross or microscopic tumor present postoperatively (i.e. positive resection margins), tumor spill before or during surgery not confined to flank, removal of tumor in > 1 piece
•Stage IV (10%): hematogenous metastases or nodal metastases outside of abdominopelvic region (e.g. lung, liver or elsewhere beyond renal drainage system)
•Stage V (5%) bilateral renal involvement at diagnosis (but each side should be staged separately as I-IV above)
Prognosis:
Poor prognostic factors:
•anaplasia in stage II-IV tumors
•high stage (most epithelial-predominant tumors are stage I;
•most blastema-predominant tumors are stage III/IV)