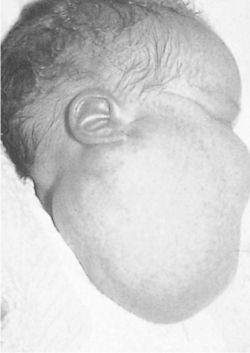
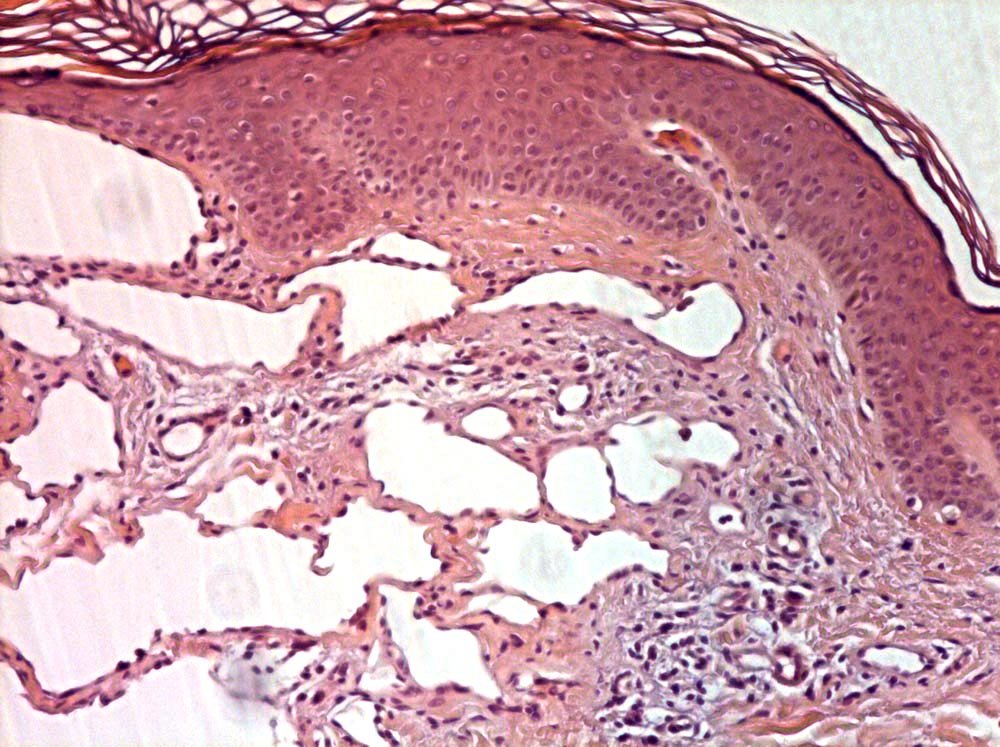
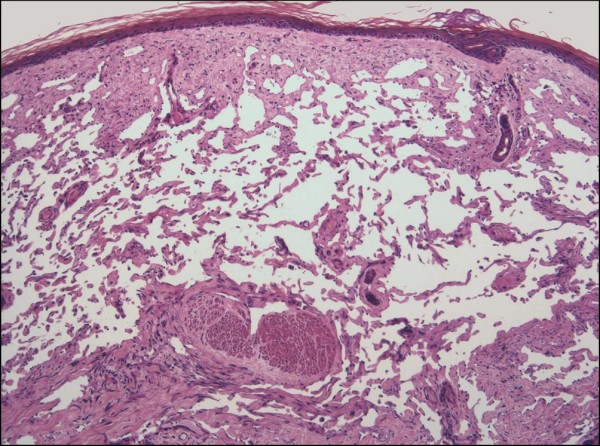
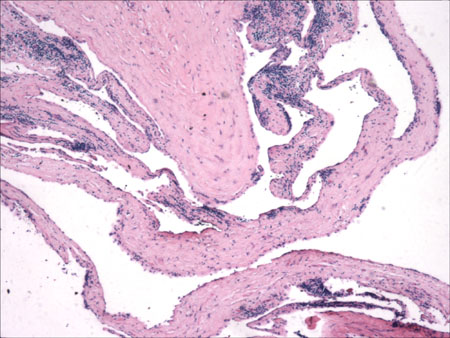
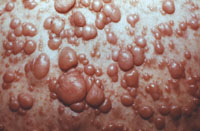
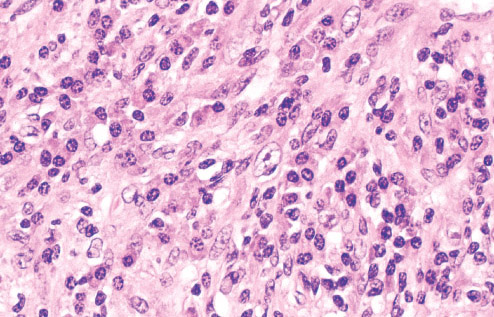
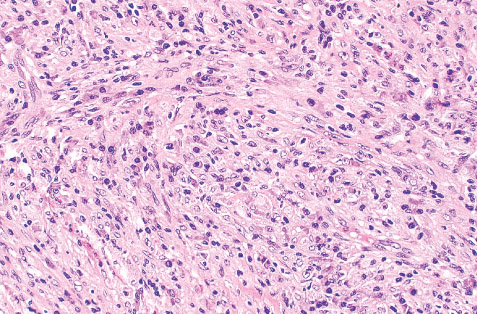
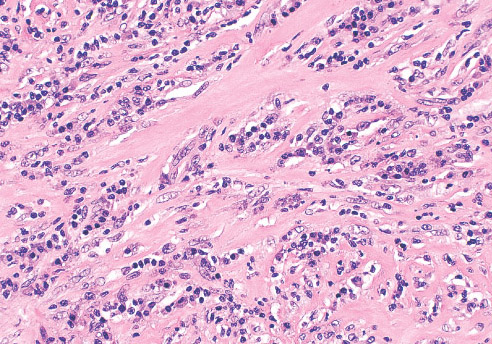
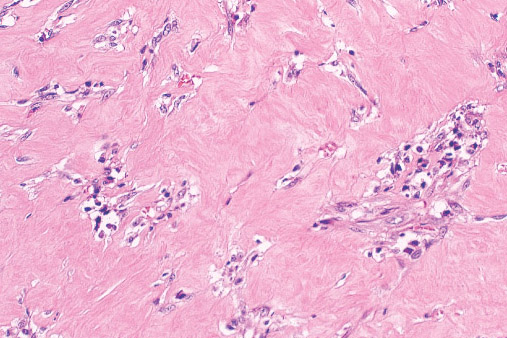
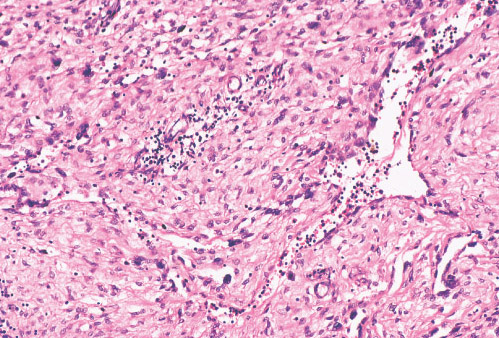
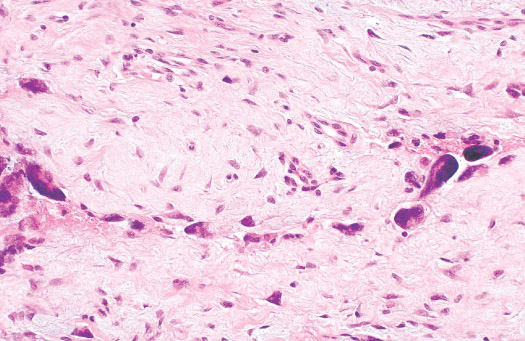
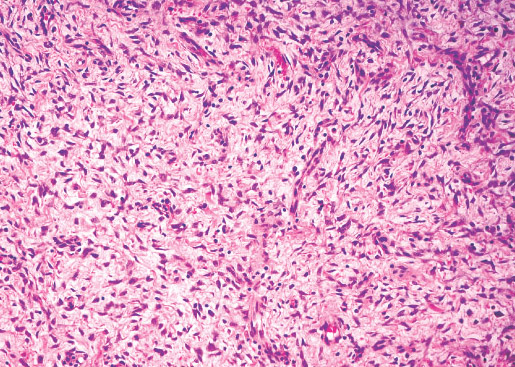
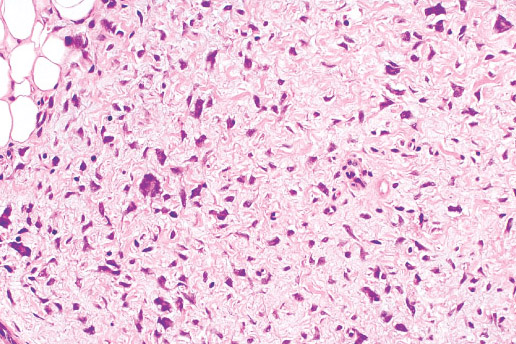
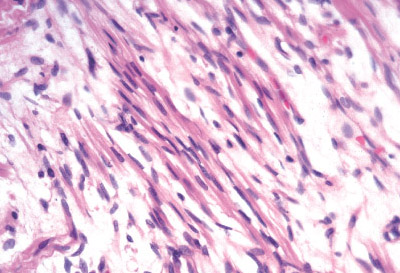
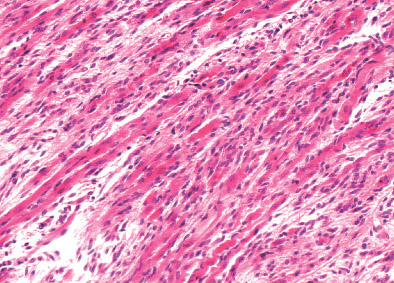
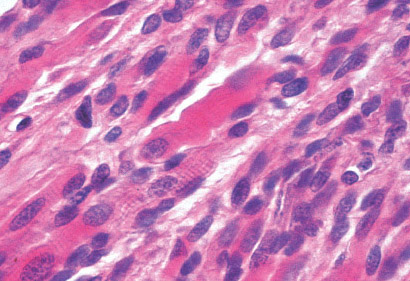
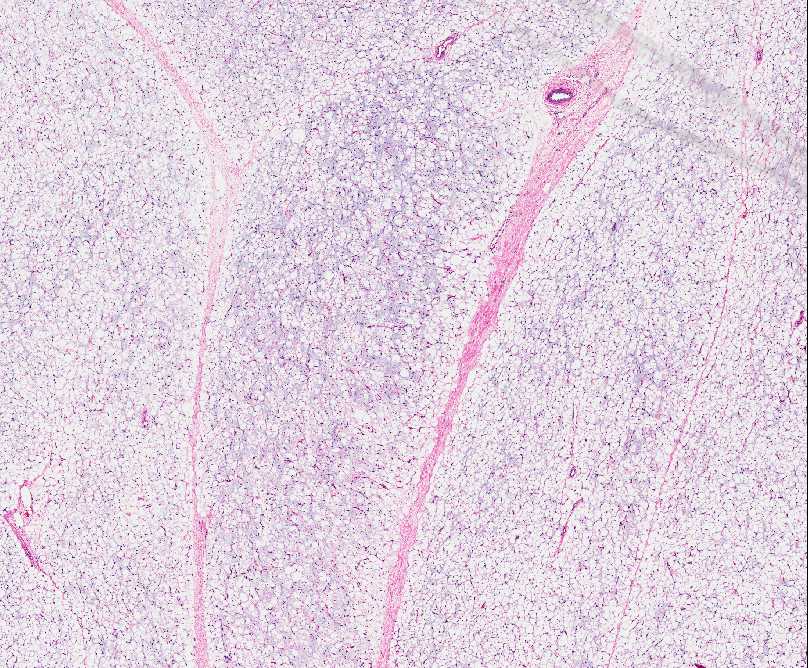
INFANTILE (JUVENILE) HEMANGIOMA
Definition:
• hemangioma which occurs during infancy.
Epidemiology:
• multiple.
Sites of involvement:
• but is most common in the region of the head and neck,
particulary the parotid, where follows distribution of
cutaneous nerves and arteries.
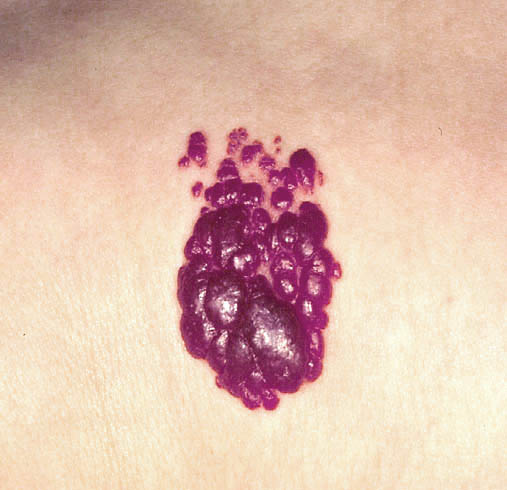
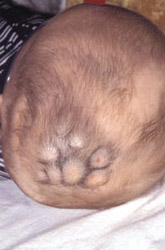
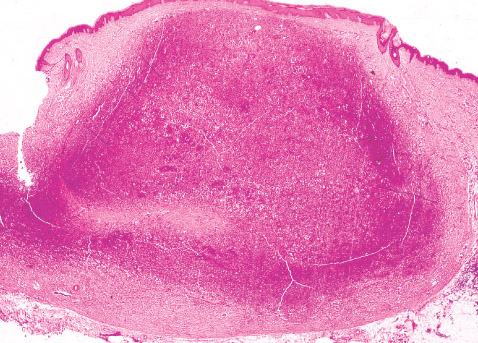
Clinical findings:
• that it is a flat, red lesion that intensifies in color when infant
strains or cries.
• distinguished it from birthmarks and sometimes is called
strawberry nevus.
• enlarge over a period of several months, achieving the
largest size in about 6-12 months; they regress over the
period of a few years. Regression show changes from
scarlet to dull grey-red and wrinkling of the skin.
• involuted, leaving small pigmented scar.
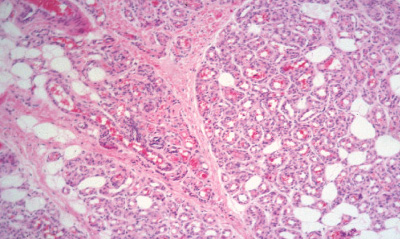
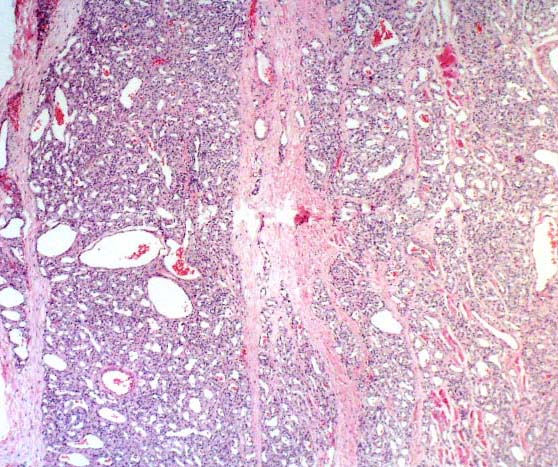
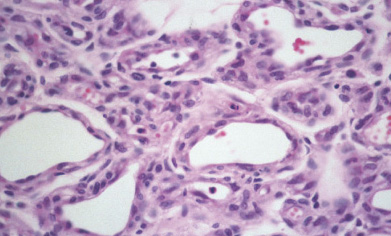
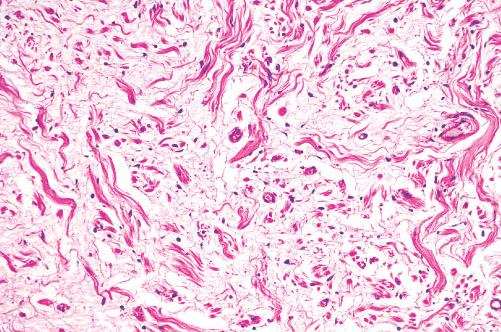
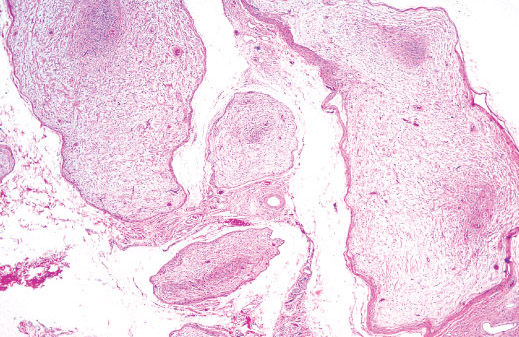
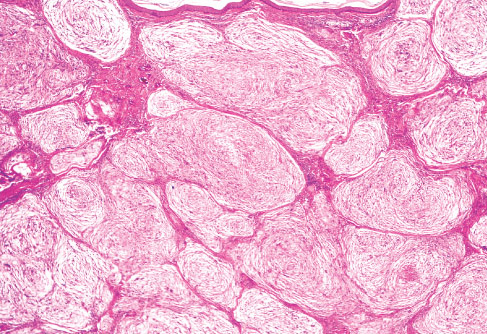
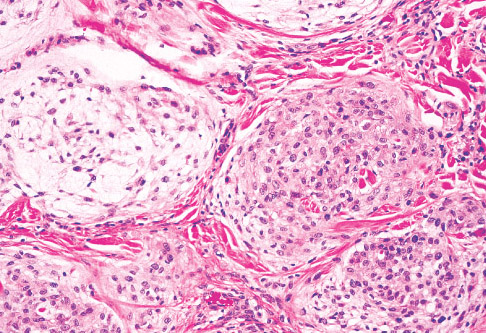
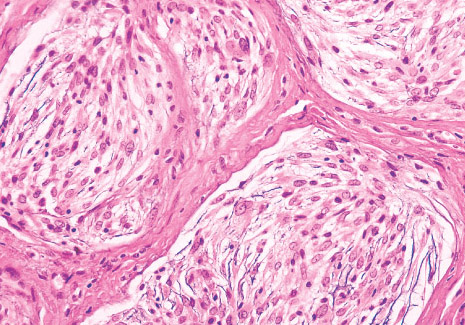
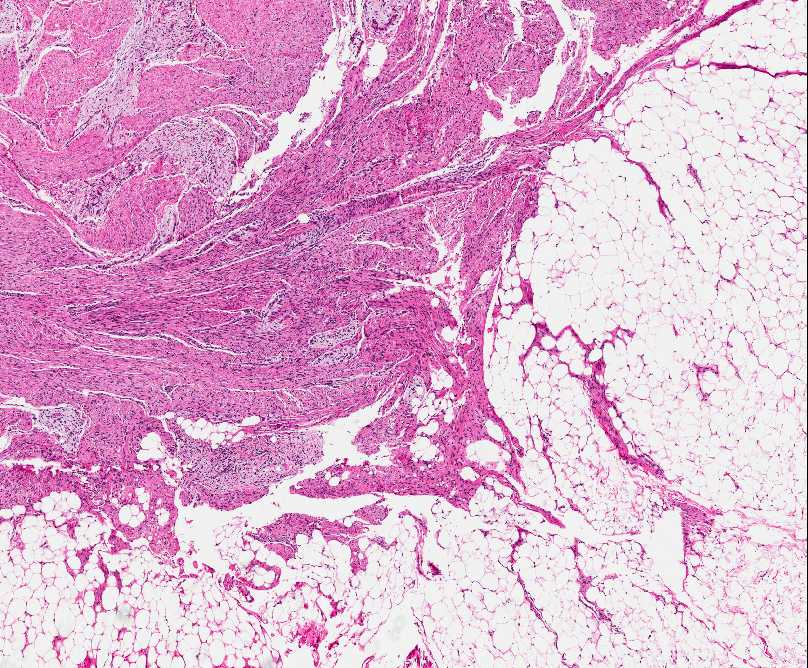
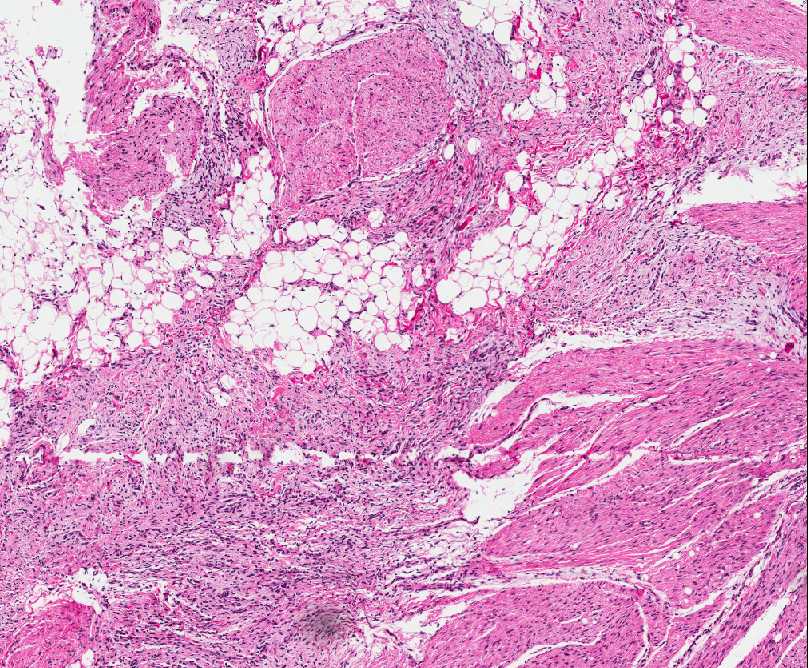
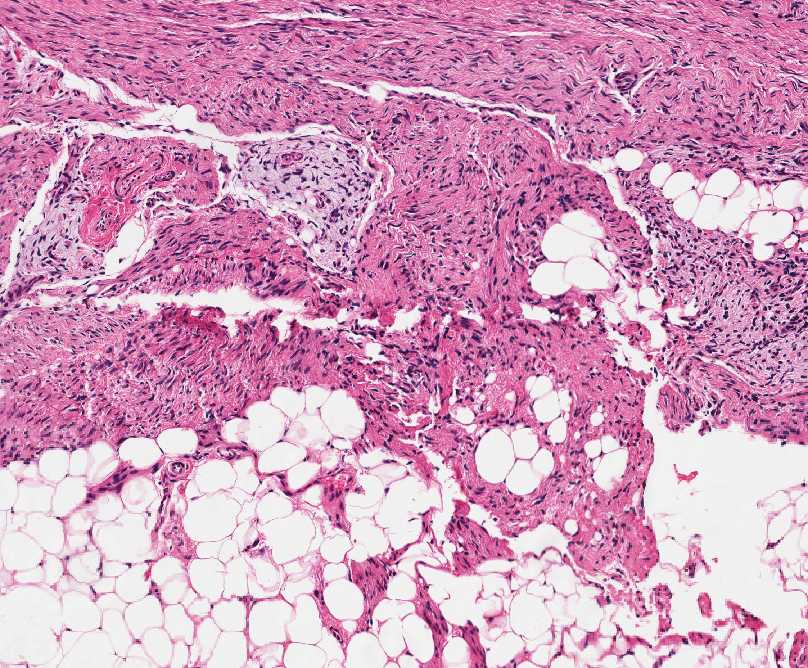
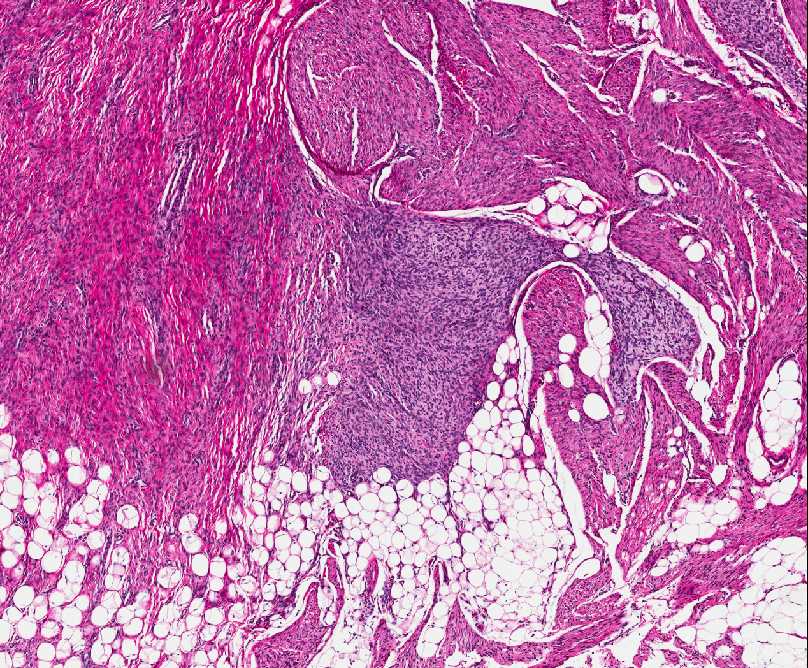
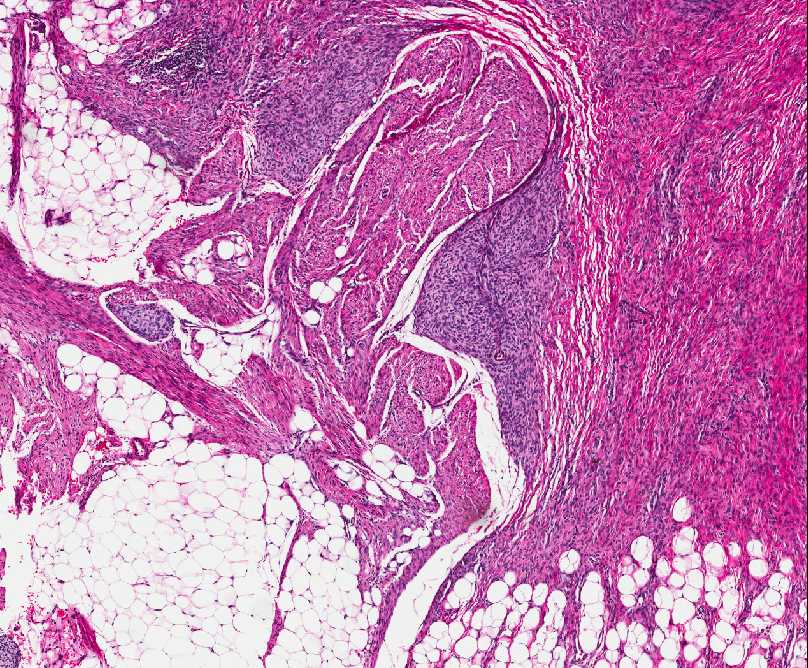
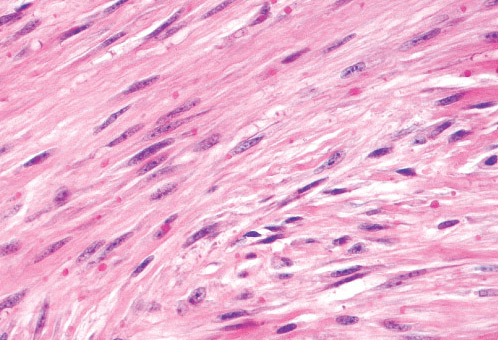
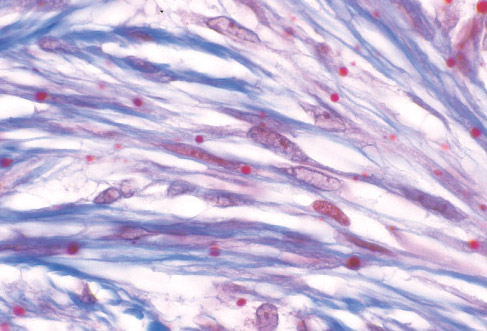
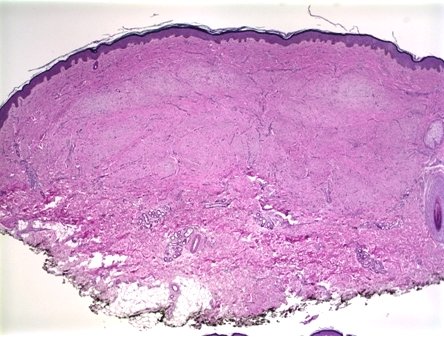
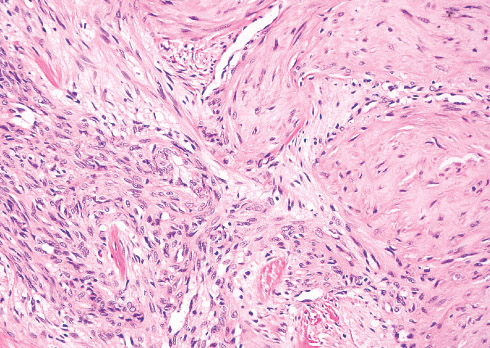
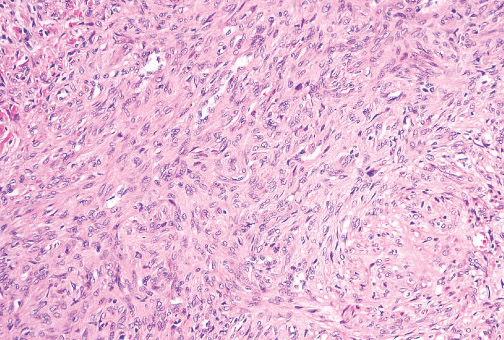
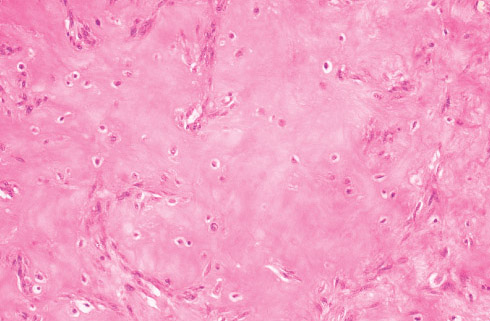
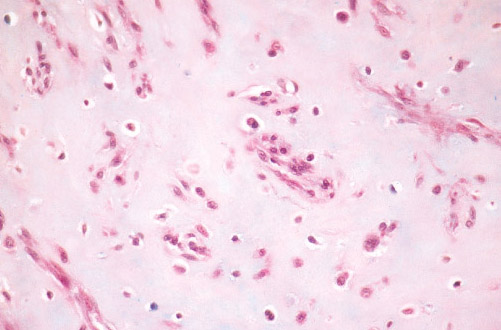
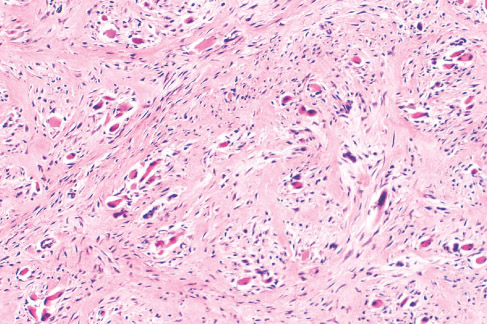
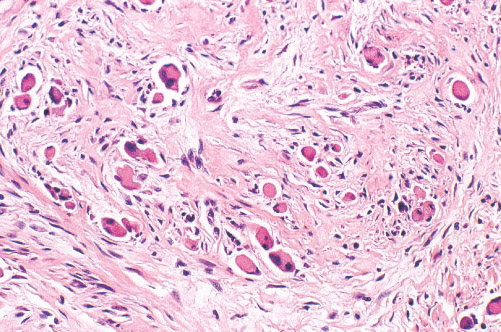
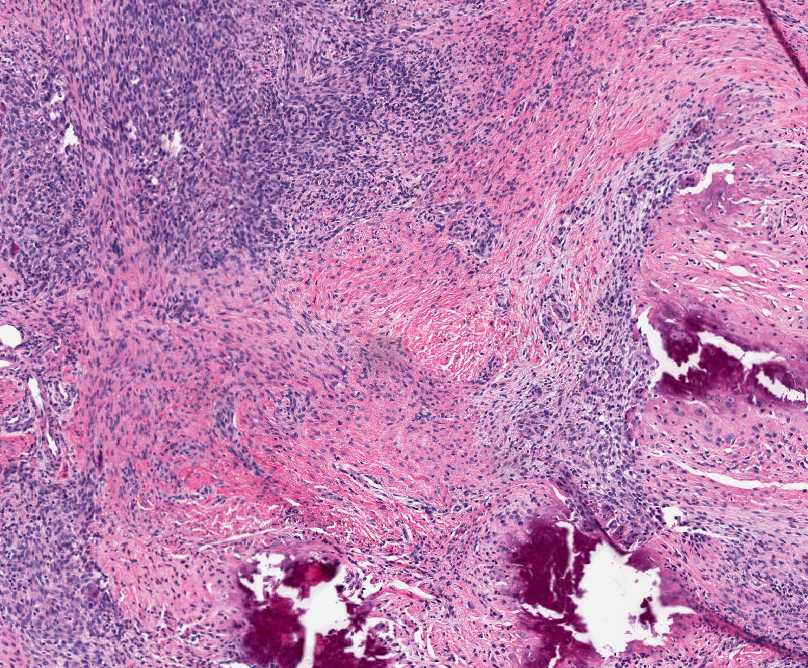
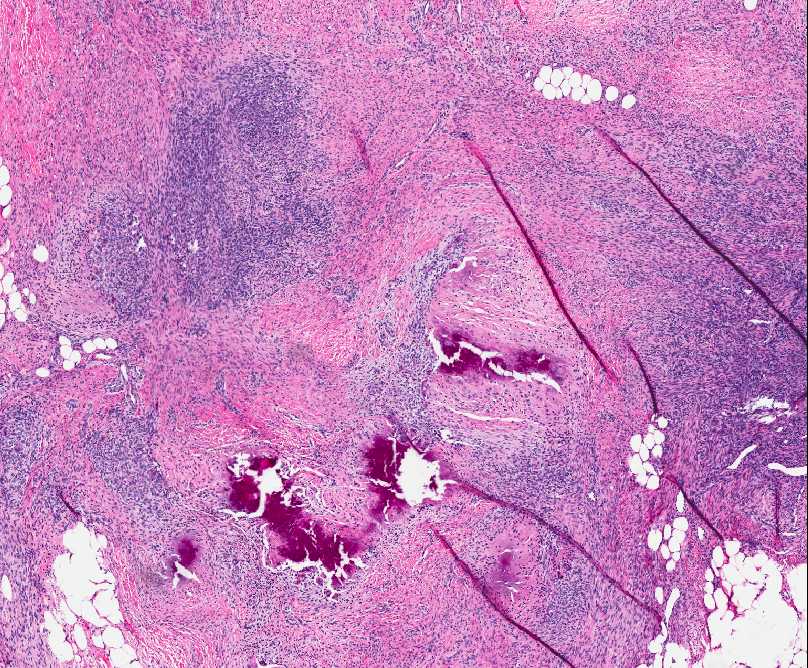
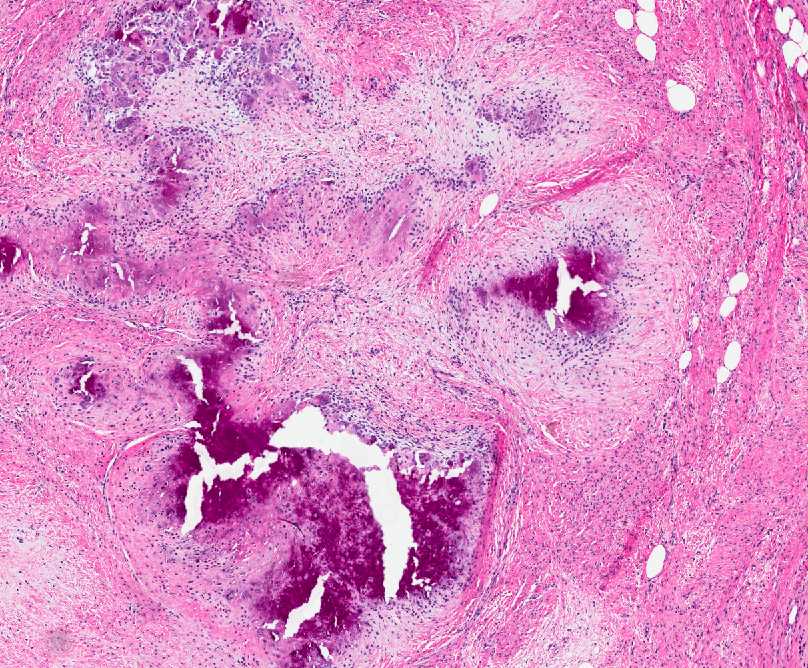
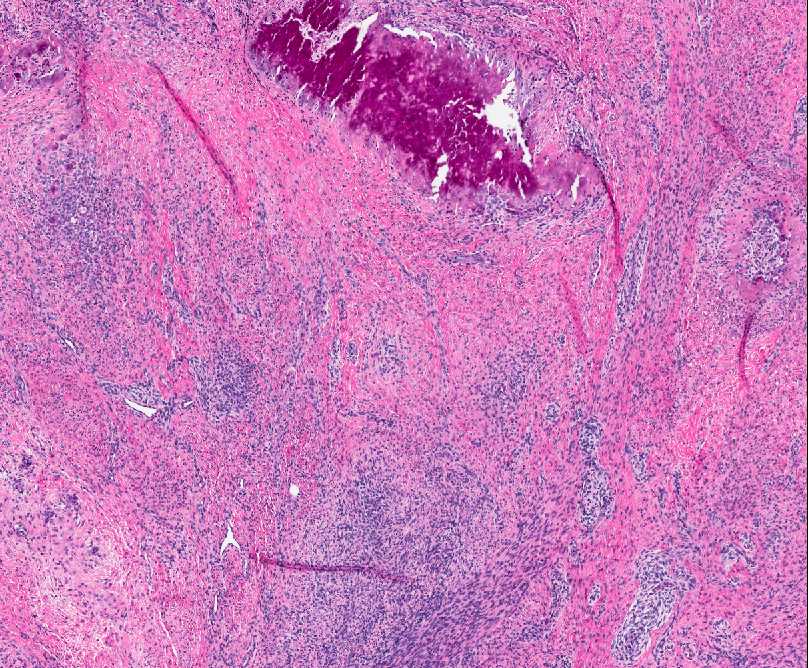
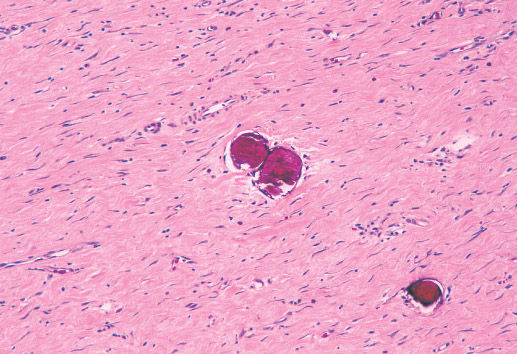
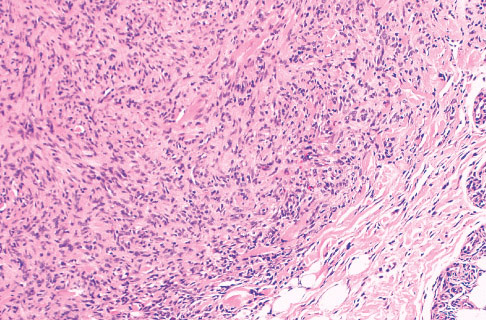
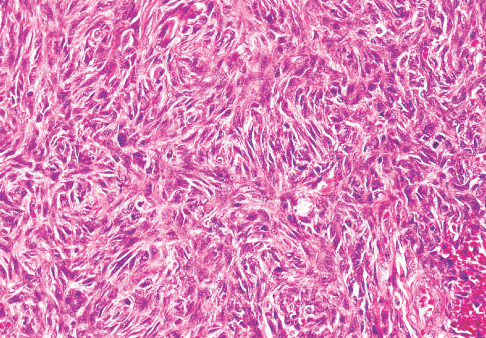
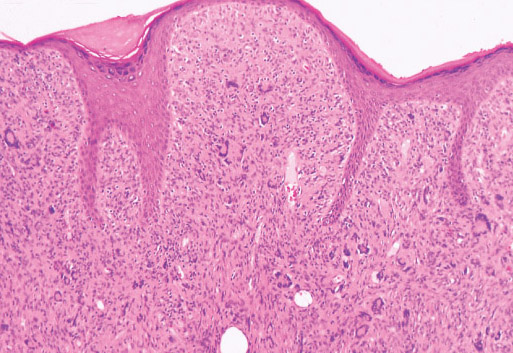
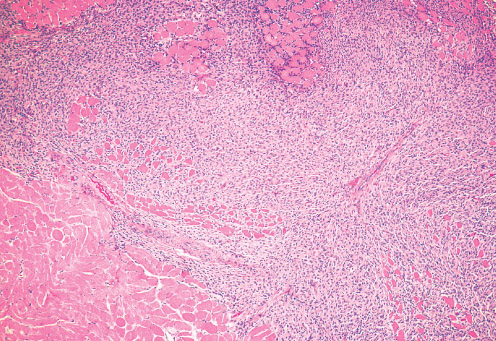
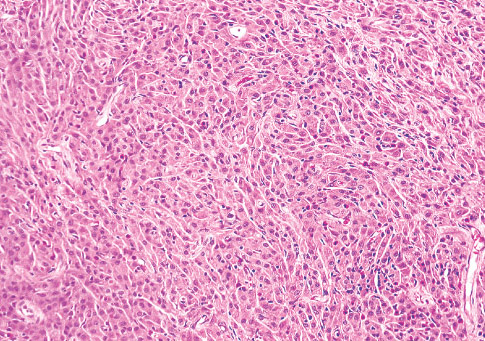
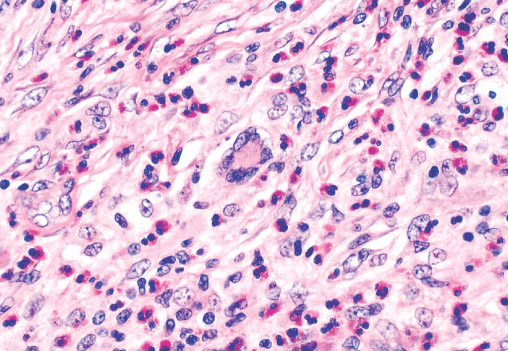
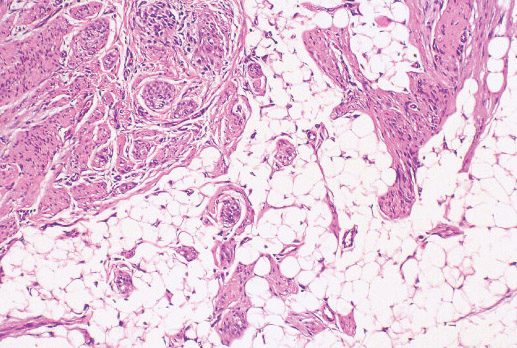
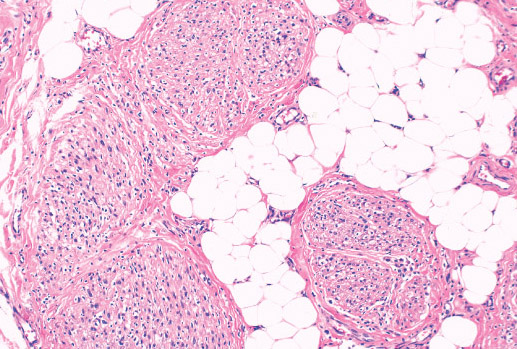
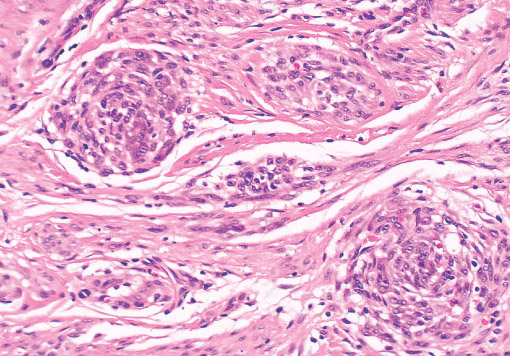
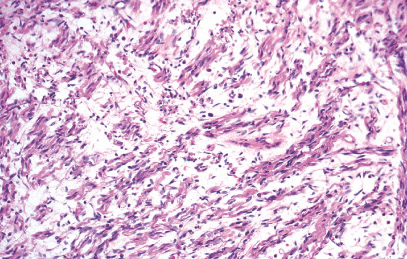
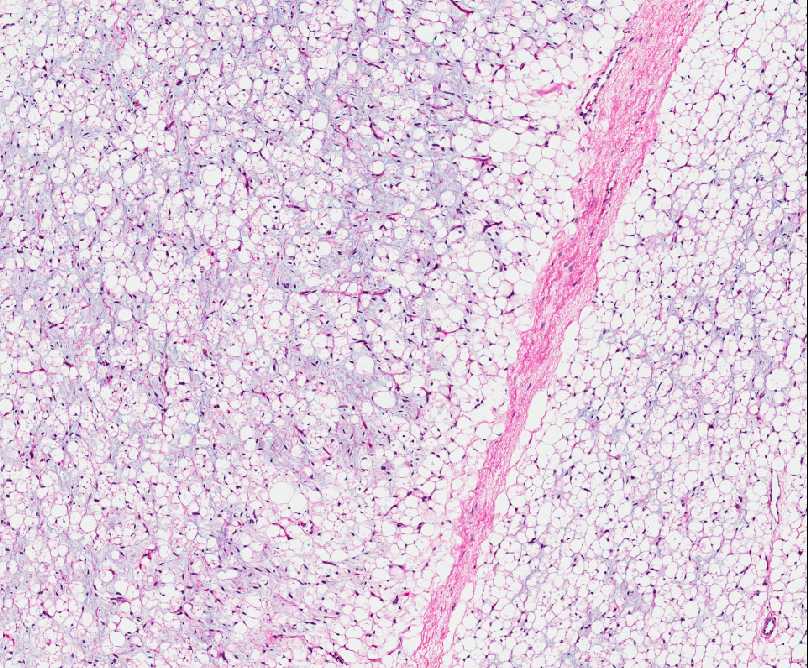
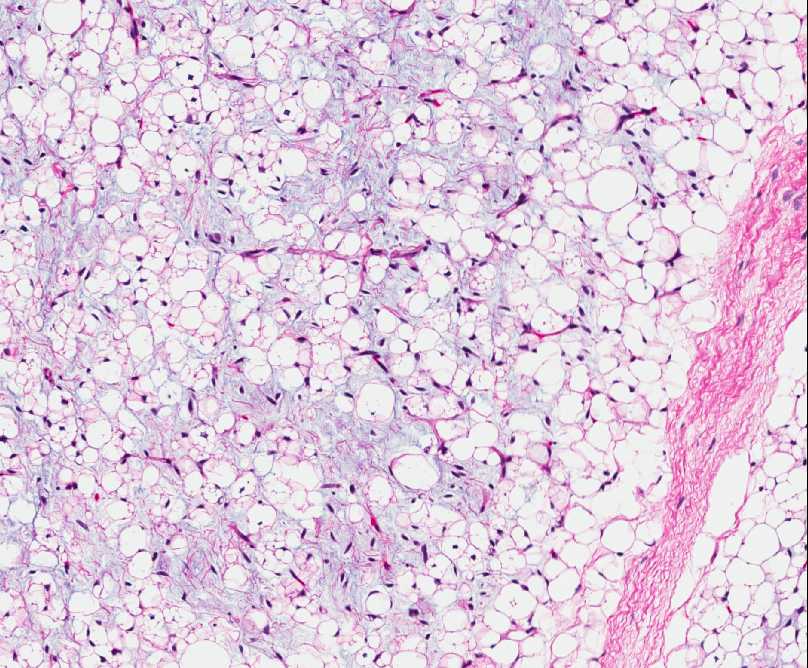
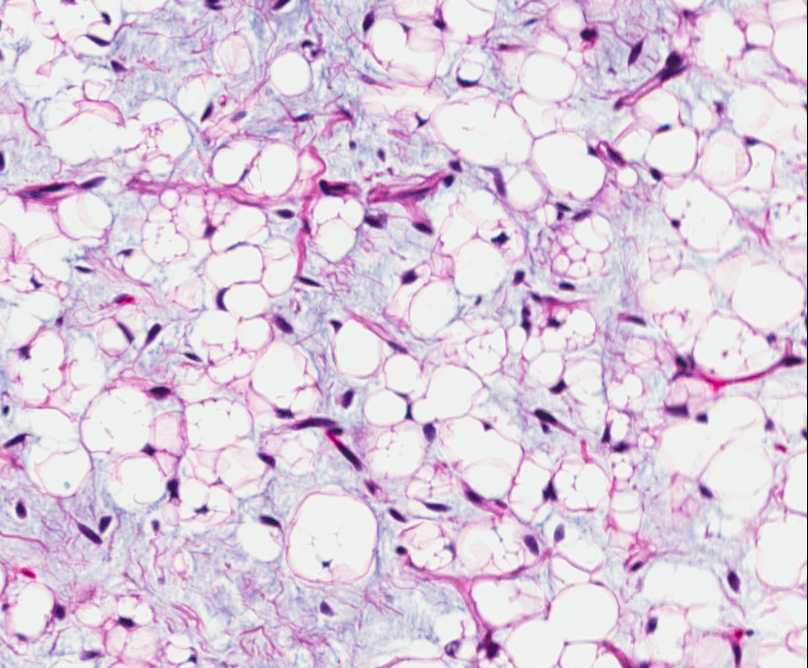
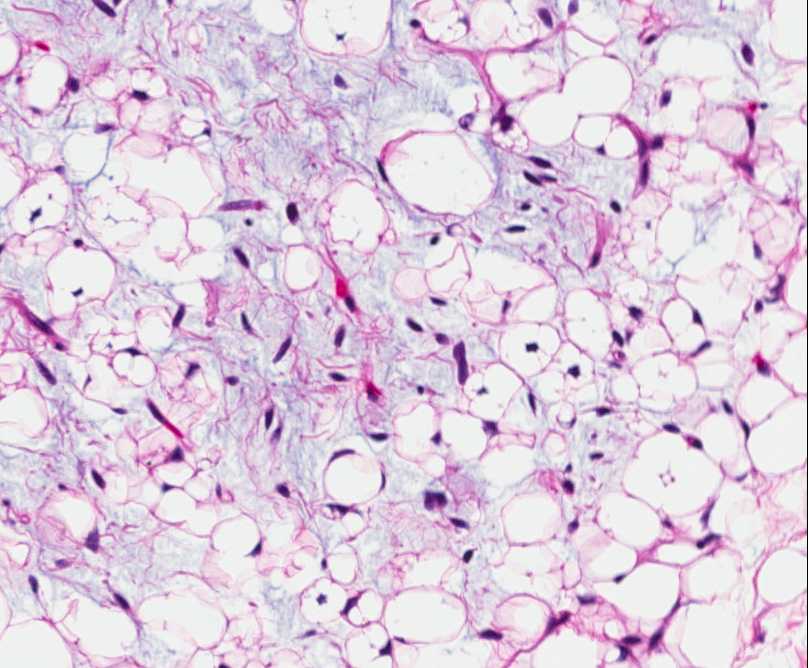
Histopathology:
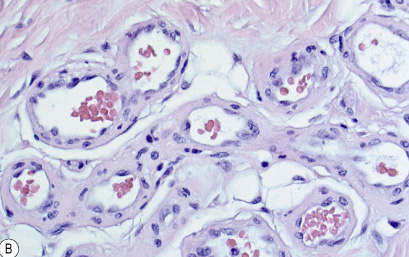
• occuring arteriole.
• • that line vascular spaces with small inconspicuous lumens.
• • • commences, the endothelium becomes flattened and
resembles adult form of capillary hemangioma.
• ultimately involves all zones.
• interstitial fibrosis.
Immunohistochemistry:
• infantile hemangioma:
• proliferative cell nuclear antigen (PCNA),
• • CD31 and vWF may be lost when lesion is
• Prognosis:
• depends on location and rate of growth.
• (e.g. airways) are usually treated with glucocorticoids until
a clinical response is achieved.
• involuted, 75-90% of cases at age 7 years.
BENIGN SOFT TISSUE TUMORS
VASCULAR TUMORS
PERIPHERAL NEUROGENIC TUMORS
FIBROUS TUMORS
FIBROHISTIOCYTIC TUMORS
MYOGENOUS TUMORS:
ADIPOSE AND MYXOID TUMORS:
OTHER
REFERENCES:
WHO Pathology and Genetics of Tumors of Soft Tissue and Bone, Lyon: IARC Press, 2002
Dorfman H. D., Bone Tumors, New York: Mosby, 1998
Potter's, Pathology of the fetus, infant and child, Mosby/Elsevier 2007
Weiss W.S., Soft Tissue Tumors, Mosby/Elsevier 2008
ANGIOMATOSIS
Definition:
• tissue occuring in infants.
Epidemiology:
• when the limb buds to form, grow proportionately with the
fetus and involve large areas of extremities and trunk.
• year of life is called diffuse neonatal angiomatosis)
Sites of involvement:
• involvement of multiple tissues (e.g., subcutis, muscle, and
bone) extensive involvement of tissue of the same type
(e.g., multiple muscles).
• occur throughout the body.
Clinical findings:
• with pain and discoloration.
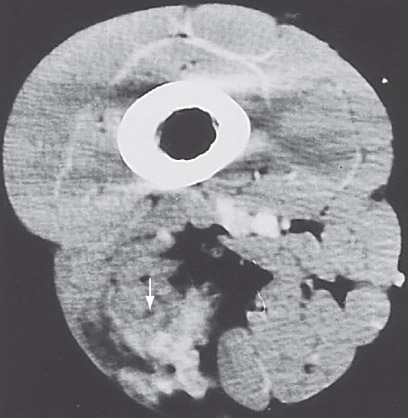
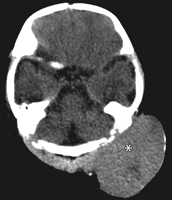
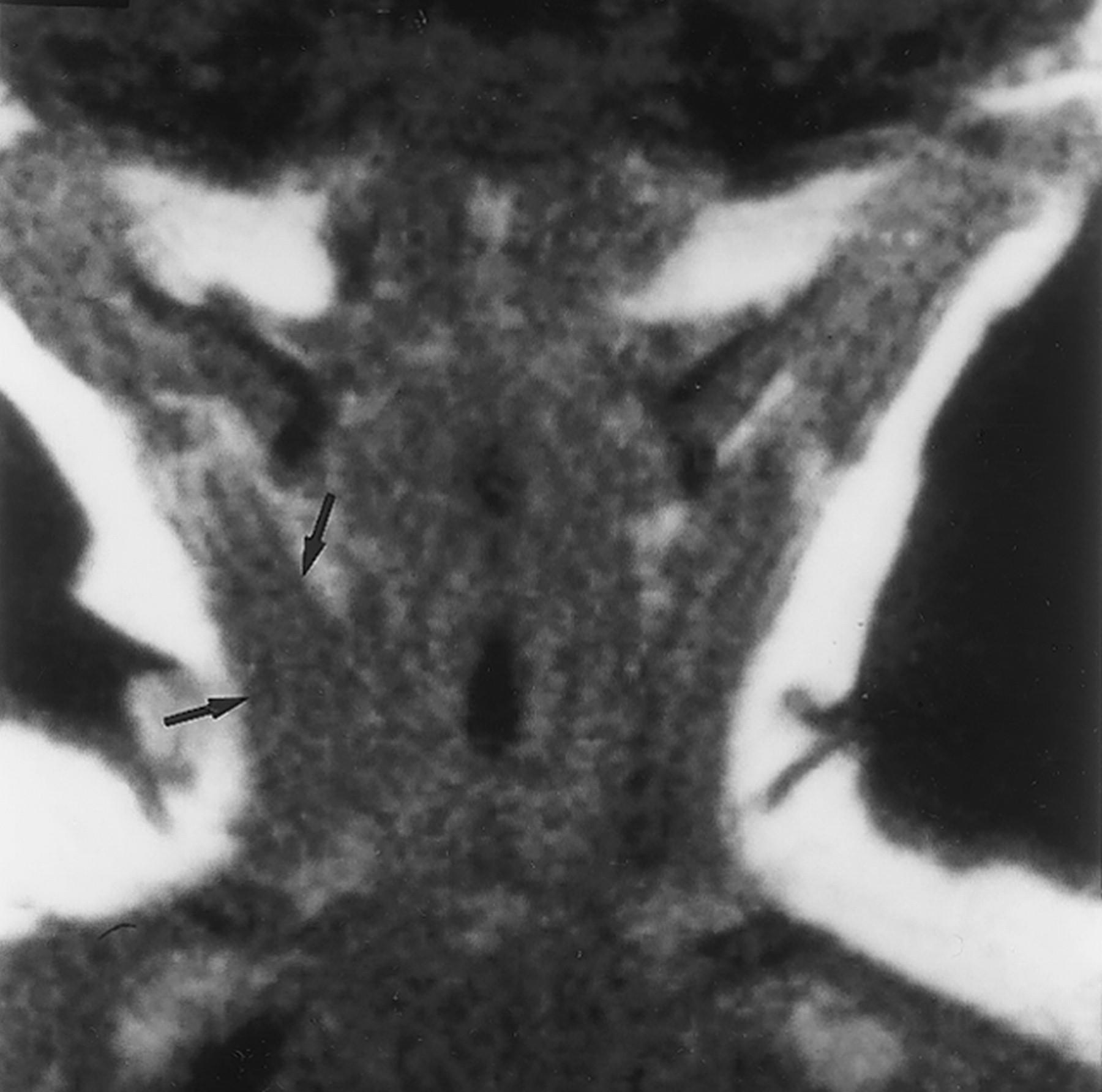
Imaging:
• may resemble sarcoma exept for presence of dense areas
corresponding to thick-walled vessels.
• Histopathology:
• • vessels of different size, composed of large venous,
cavernous, and capillary-sized vessels scattered in soft and
fat tissue.
• walls that have occasional attenuations and herniations.
• vessels clustered around the wall of large vein.
• hemangiomas, exept that the nodule of tumor diffusely
infiltrate the surrounding soft tissue.
• the alternative designation of "infiltrative angiolipoma".
Prognosis:
• had more than one recurrence within
•
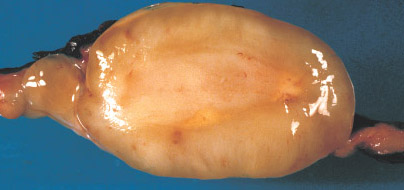
LYMPHANGIOMA
Definition:
• dilated lymphatic channels.
Epidemiology:
• present at birth or during first year of life.
• chromosome X, 46 X) and may be found in abortuses.
• the most common type and is also called cystic hygroma.
Sites of involvement:
• and groin.
• trunk, limbs and abdominal sites including mesentery and
retroperitoneum.
Clinical features:
• which are soft and fluctuant in palpation, and can show
displacement surrounding organs at mediastinal or abdominal
sites.
Imaging:
• vascularization and CT scan reveals multiple, homogenous,
nonenhancing areas.
Gross:
• up of one or more large interconnecting cysts to ill-defined,
sponge-like compressible lesion composed of microscopic
cysts.
• lymphangiomas (cystic hygroma) and the latter as
cavernous hemangioma.
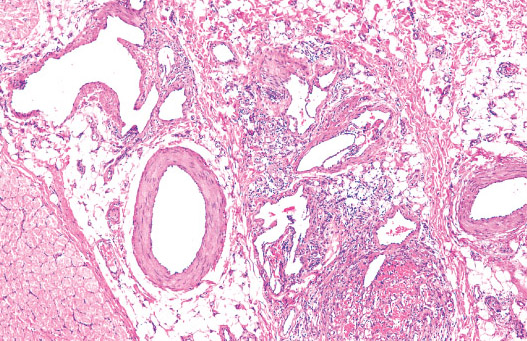
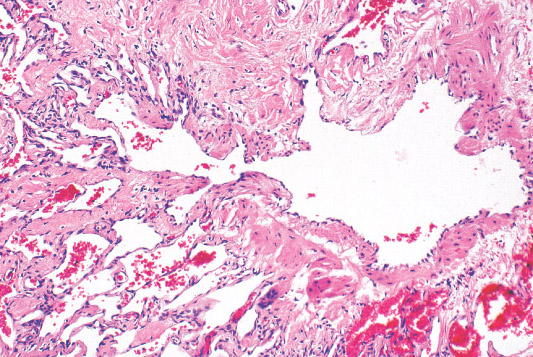
Histopathology:
• dilated lymphatic vessels of different size, which are lined by
a flatened endothelium and frequently surrounded by
lymphocytic aggregates.
• fluid, lymphocytes and sometimes erythrocytes.
• and long standing lesions may show fibrosis and
inflammatory changes.
• is frequently seen.
Immunohistochemistry:
• Prognosis:
• malignat transformation does not occur.
• and may compromise trachea, esophagus.
PERIPHERAL NEUROGENIC TUMORS
NEUROFIBROMA:
Definition:
• tumor consisting of mixture of cell types, including Schwann
cells, perineural-like cells, and fibroblasts; multiple and
plexiform neurofibromas are typically associated with
neurofibromatosis type 1.
Epidemiology:
• solitary nodules unrelated to any apparent syndrome or as
solitary, multiple or numerous lesions in individuals with
neurofibromatosis type 1 (NF1 loss of 17q gene region).
• Sites of involvement:
• (localized cutaneous neurofibroma) and less often as a
circumscribed mass in a peripheral nerve (localized intraneural
neurofibroma) or as a plexiform enlargement of a plexus or
major nerve trunk.
Clinical features:
• • in which they are associated with pigmented cutaneous
macules (cafe-au-lait spots) as well as 'freckling', often axillary
in location.
Gross:
• rather circumscribed, or are diffuse and involve skin and
subcutaneous tissue.
• • ("bag of warms") when tumor involves multiple trunks of a
plexus or rope-like lesions when multiple fascicles of a large,
non-branching nerve such as the sciatic are affected.
Histopathology:
• thin, curved to elongated nuclei and scant cytoplasm as well as
fibroblasts in a matrix of collagen fibers and Alcian blue-positive,
myxoid material.
• schwannomas.
• neurofibroma) or significantly increased cellularity (cellular
neurofibroma).
• • looks like dense, retractile bundles resembling "shredded
carrots".
• nerve fibers, which become enmeshed by tumor.
• remain confined to the nerve and encompassed by it thickening
epineurium.
• diffusely into the surrounding dermis and soft tissue.
• lack hyalinization.
Prognosis:
• considered a precursor lesion to the majority of malignant
peripheral nerve sheath tumors.
• tumors, but is rare event in diffuse cutaneous and massive soft
tissue neurofibromas.
• with NF1 and should be investigated for other evidence of the
disorder.
NEUROTHECOMA
Definition:
• myxoma.
Epidemiology:
• Sites of involvememt:
• tissue.
• such as the head, neck, and shoulder.
Clinical findings:
• Histopathology:
• due to fibrous bands divinding lesion into irregular lobules.
• stroma, so tumor may appear myxoid or solid.
• atypia or mitotic activity.
• neurites are identified among the tumor cells.
• cellular, with marked cellular atypia, rare mitoses and
extension to adjacent soft tissue ( cellular neurothecoma),
which may be mistaken for sarcoma.
• atypical features are the superficial dermal location and the
distinctive septate architecture.
Immunohistochemistry:
• Prognosis:
•
Please email me your comments about the site.
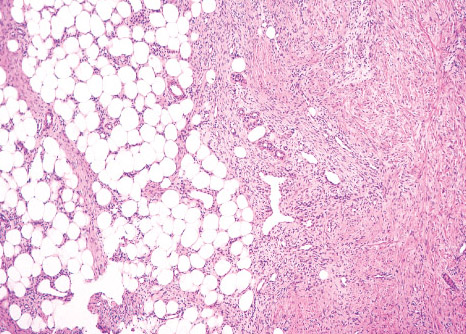
FIBROUS HAMARTOMA OF INFANCY
Definition:
• mass with three components: dense fibro-collagenous tissue,
loosely textured areas of immature rounded mesenchymal
cells and mature fat.
Epidemiology:
• of the relatively more common tumors of fibrous tissue in early
childhood.
Sites of involvement:
• followed by the upper arm and shoulder, thigh, groin, back,
and forearm.
• Clinical features:
• first 2 years of life and up to 25% are discovered at birth.
• predominance.
• lesion, rapidly growing, freely movable mass in the subcutis
or dermis, occasionally attached to underlying fascia and
rarely involving skeletal muscle.
Gross:
• with yellow fat. Most lesions are less than 5 cm in diameter,
tumors rarely reach larger than 10 cm.
Histopathology:
• structures.
• fibrocollagenous tissue are composed of fibroblastic and
myofibroblastic spindle cells with bland, straight or wavy
nuclei separated by varying amounts of collagen.
• small, rounded or stellate, primitive mesenchymal cells with
scant cytoplasm embedded in myxoid matrix containing
abundant hyaluronidase-sensitive acid mucopolysaccharides.
• small veins.
• two components.
• cases.
Prognosis:
• local excision.
•
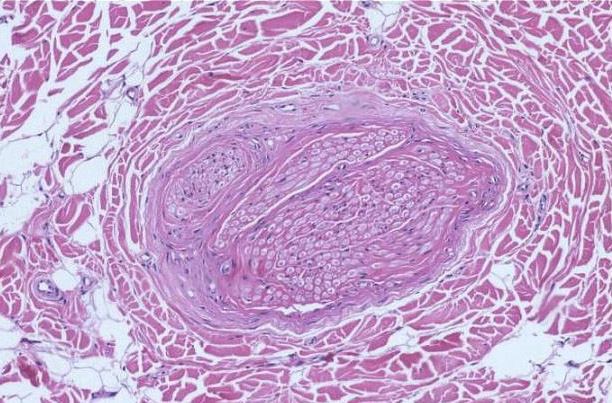
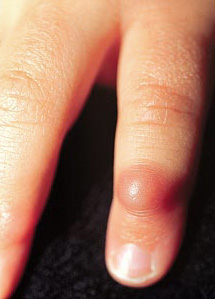
INFANTILE DIGITAL FIBROMATOSIS (INCLUSION BODY FIBROMATOSIS)
Definition:
• that typically occur on the digits of young children. It is named
for the intracytoplasmic round inclusions staining red with
trichrome.
Epidemiology:
• Sites of involvement:
• hands and feet.
• • • sites such as the soft tissue of the arm or breast.
Clinical features:
• • • the overlying skin is typically taught and strched.
Gross:
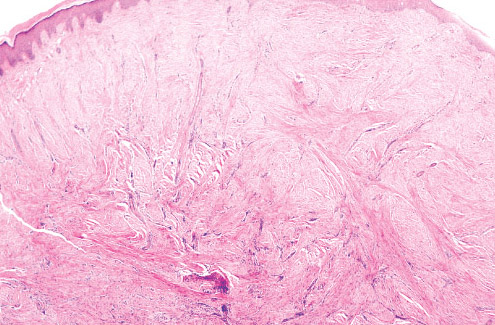
• • Histopathology:
• of uniform spindle cells associated with varying amounts of
extracellular collagen.
• adjacent soft tissue. Individual cells have central elongated
nuclei and vaguely fibrillar cytoplasm.
• spherical eosinophilic "inclusion". Inclusions are brightly red
trichrome positive and PAS negative.
• uniformly distributed.
• prominent mitoses present.
Prognosis:
• • •
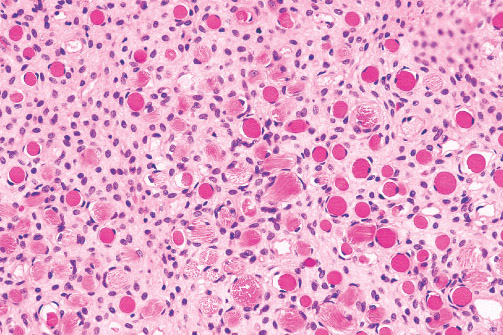
MYOFIBROMA/MYOFIBROMATOSIS
Definition:
• benign neoplasm composed of contractile myoid cells arranged
around thin-walled vessels.
• myopericytoma and so-called infantile hemangiopericytoma.
Epidemiology:
• diagnosed at birth or within the first two years of life.
• • unclear.
• Sites of involvement:
• cutaneous/subcutaneous tissues of head and neck region,
followed by trunk, lower and upper extremities.
• small number involving bone, predominantly the skull.
• bone and frequently (10-20%) occurs in the deep soft tissue
and visceral location.
Clinical features:
• • simulating vascular neoplasm.
• movable masses while more deeply seated lesions may be
fixed.
• that are involved.
Imaging:
• infiltrative, often with calcifications, either within or surrounding
the lesion.
• radiolucent lesions within the metaphyseal regions, spering
the region adjacent to epiphysis.
Gross:
• • than those in deep soft tissue.
• brown.
• filled with caseous-like material or hemorrhage.
Histopathology:
• proliferation with zonation.
• myofibroblasts arranged in short fascicles or whorls.
• have elongated, tapering nuclei with vesicular chromatin pattern
and one or two nuclei.
• • they are round, polygonal or spindle cells with slightly larger
hyperchromatic nuclei.
• thin-walled, irregulary branching, hemangipericytoma-like
vessels.
• • frequently.
• Immunohistochemistry:
• S100.
Prognosis:
• • suggesting
• • prognosis, with involvement of vital organs, leading to
cardiopulmonary or gastrointestinal complication, causing
death in rare cases.
• factor. out 50% of cases.
• •
JUVENILE HYALIN FIBROMATOSIS
Definition:
• extracellular "hyaline material" within skin, somatic soft tissues
and the skeleton mimicking tumor like masses, and typically
presents in infancy.
Epidemiology:
• • • • and size of superficial and deep nodules with resulting deformity
and dysfunction.
Sites of involvement:
• (particulary the face and neck resulting in papules and nodules),
gums (causing gingival hyperplasia), periarticular soft tissue
(resulting in joint contractures) and bones (especially the skull,
long bones and phalanges).
Clinical features:
• in particular around the ears.
• • contracures, most commonly involving knees and elbows.
Imaging:
• osteoporosis and discrete lytic lesions.
Histopathology:
• • with extracellular uniform hyaline material (produced by
fibroblasts) that is non-fibrillar and eosinophilic in H&E stain.
• more cellular.
• fascicular arrangement.
• • compressed.
• Prognosis:
• location.
• • and the patient's functional impairement.
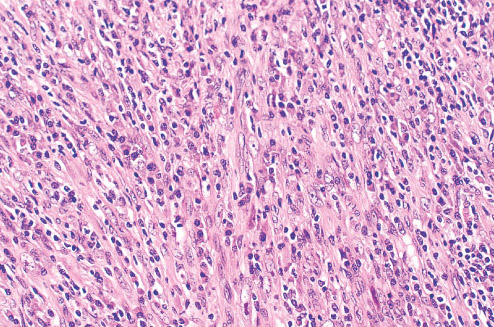
FIBROMATOSIS COLLI
Definition:
• sternocleidomastoid muscle of infants.
• cervico-fascial assymetry due to its shortening (torticollis).
Epidemiology:
• • • Sites of involvement:
• muscle.
Clinical features:
• the distal sternocleidomastoid muscle.
Imaging:
• Gross:
• Histopathology:
• conventional fibroma.
• myofibroblastic cells embedded in rich collagen.Infiltration and
entrapment of skeletal myocytes is evident.
Prognosis:
• • • demonstrate normal cervico-fascial posture and movement with
this approach.
• of patients.
• older than 1 year.
INFANTILE FIBROMATOSIS, LIPOFIBROMATOSIS
Definition:
• extremities.
Clinical features:
• rarely occurs in the thigh, trunk and head.
• • • Gross:
• Histopathology:
• reflecting progressive stages in the differentiation of the
fibroblasts.
• (mesenchymal) type. This form is usually found in infants in first
few months of life and is characterized by small, haphazardly
arranged, round or oval cells deposited in a myxoid background.
The cells are intermediate in appearance between primitive
mesenchymal cells and fibroblasts and they are intimately
associated with residual muscle fibers and lipocytes. Some
cases have extensive lipocytic elemts, and is reffered as
lipofibromatosis.
Peripherally located lymphocytic inflammation is often present.
Sometimes tumor may be highly cellular and mitotically active,
making distinction from infantile fibrosarcoma difficult.
• virtually indistinguishable from the adult form of fibromatosis
(desmoid tumor). This type usually occurs in children older than
5 years of age and behaves like adult desmoid tumor. Although
the morphology is similar to aduilt lesion, calcification and/or
ossification is a feature peculiar to pediatric cases.
Immunohistochemistry:
• actins and EMA.
Prognosis:
• metastatic potential.
NFLAMMATORY MYOFIBROBLASTIC TUMOR
Definition:
• distinctive lesion composed of myofibroblastic spindle cells
accompanied by infiltrative inflammatory component of plasma
cells, lymphocytes and eosinophils.
• young adults.
Epidemiology:
• young adults.
• • • although no exact etiology is known.
Sites of involvement:
• are the lung, mesentery, and omentum.
Clinical findings:
• • be asymptomatic.
• • growth failure, malaise, weight loss, anemia, and elevated
erythrocyte sedimentation rate.
• reapperance may be sign of recurrence.
Imaging:
• • Gross:
• whorled fleshy or myxoid cut surface.
• minority of cases.
Histopathology:
• three basic histological patterns of IMT.
• myofibroblasts in an oedematous myxoid background with
abundant blood vessels and an infiltrate of plasma cells,
lymphocytes and eosinophils resemble granulation tissue,
nodular fasciitis, or other reactive processes.
• spindle cell proliferation with variable myxoid and collagenized
regions and a distinctive inflammatory infiltrate with diffuse
inflammation, small aggregates of plasma cells or lymphoid
nodules.This may resemble a fibromatosis, fibrous
histiocytoma, or a smooth muscle neoplasm. In some
instances, the spindled myofibroblastic cells surround blood
vessels or bulge into vascular spaces, similar to infantile
fibromatosis or intravascular fasciitis. Ganglion-like
myofibroblasts with vesicular nuclei, eosinophilic nucleoli, and
abundant ampophilic cytoplasm are often seen these two
patterns.
• with pale-like collagen, lower cellularity, and relatively sparse
inflammation with plasma cells and eosinophils. Coarse or
psammomatous calcifications and osseous metaplasia are
occasionally seen.
Immunohistochemistry:
• (smooth muscle actin).
Genetics:
• ALK receptor tyrosine kinase gene in chromosome band 2p23.
Prognosis:
• location, resectability and multinodualrity.
• response to corticosteroids and NSAID
CALCIFYING APONEUROTIC FIBROMA
Definition:
• for local recurrence.
• characteristic for this lesion.
Epidemiology:
• • • Sites of involvement:
• • • growing, poorly circumscribed non-tender mass.
Gross:
• Histopathology:
• • of rounded, chondrocyte-like cells, arranged in short, parallel
arrays.
• coalescent calcified nodules emanating into the surrounding soft
tissue.
• features.
• Immunophenotype:
• • • of diagnosis.
• likelihood of recurrence is not predictable on the basis of
morphology, location or the completeness of the primary
excision.
• CALCIFYING FIBROUS PSEUDOTUMOR (CALCIFYING FIBROUS TUMOR)
Definition:
• • psammomatous and dystrophic calcification, and patchy
lymphoplasmacytic infiltrates.
Epidemiology:
• gender predilection.
Sites of involvement:
• (extremity, trunk, neck and scrotum) but have been reported all
over the body.
Clinical features:
• • Imaging:
• • punctate.
• appearance and a signal closer to that of muscle than fat.
Gross:
• from <1 to 15 cm.
Histopathology:
• paucicellular, hyalinized fibrosclerotic tissue with a variable
inflammatory infiltrate consisting of lymphocytes and plasma
cells.
• • throughout.
Immunophenotype:
• Prognosis:
• BENIGN FIBROHISTIOCYTIC TUMORS
BENIGN FIBROUS HISTIOCYTOMA
Definition:
• histiocytic cells arranged in sheets of short fascicles and
accompanied by inflammatory cells, foam cells and siderophages,
which may develop within subcutaneous tissue, deep soft tissue
or in parenchymal organs.
• Epidemiology:
• tumor.
• • Sites of involvement:
• common sites.
• described in muscle, mesentery, trachea and kidney.
Clinical features:
• enlarging mass.
• Gross:
• from a few milimeters to few centimeters.
Histopathology:
• dermis and occasionally subcutis.
• with dermal collagen ("collagen traping").
• with elongated or plump vesicular nuclei and eosinophilic,
ill defined cytoplasm.
• may be common but usually less than 5 per 10 HPF.
• xanthoma like cells.
• but show more prominent storiform pattern and fewer secondary
elements like xanthoma cells.
Immunohistochemistry:
• Prognosis:
• JUVENILE XANTHOGRANULOMA
Definition:
• childhood.
Epidemiology:
• more cutaneous nodules and less often by additional lesions in
deep soft tissue or organs.
• are usually solitary.
Clinical findings:
• the trunck and extremities.
• • brown or yellow.
• accompanied by similar lesions in other sites, such as the eye,
lung, epicardium, oral cavity or testis.
Histopathology:
• dermis and extending to, but not invading, the flattened epidermis.
• extends into sub cutis.
• muscle at the periphery.
• pleomorphism with rare mitoses.
• • xanthomatous cytoplasm.
• • including lymphocytes, neutrophils, and eosinophils.
• storiform pattern resembling conventional fibrous histiocytoma
seen in adults.
Imunohistochemistry:
• CD31 and factor XIIIa and negative for CD1a and S100.
Prognosis:
• large , deeply localized lesions pursue a favorable course.
GIANT CELL FIBROBLASTOMA
Definition:
• back of the thigh, inguinal region and chest wall in children younger
than 5 years of age.
Epidemiology:
• medical attention, and the median age was 3 y/o. Male
predominant (75%).
Clinical findings:
• the back of the thigh, inguinal region, and chest wall.
Gross:
• measure 1-8 cm.
Histopathology:
• degree of nuclear pleomorphism that infiltrate the deep dermis and
subcutis and encircle adnexal structures in fasion similar to
dermatofibrosarcoma protuberans.
• dermatofibrosarcoma protuberans to those that are hypocellular
with a myxoid or hyaline stroma.
• by giant cells.
Immunohistochemistry:
• negative for S 100 and vascular markers.
Prognosis:
• PLEXIFORM FIBROHISTIOCYTIC TUMOR
Definition:
• dermis, occurs almost exclusively in children and young adults.
Epidemiology:
• occurring almost exclusively in children and young adults.
Sites of involvement:
• the lower extremity (14%).
Clinical features:
• Gross:
• trabecular appearance.
Histopathology:
• component and a round cell histiocytic component containing
multinucleated cells.
• nodules that occupy the dermis and subcutaneous tissue.
• contain multinucleated, osteoclast-like giant cells and occasionally
undergo focal hemorrhage. The cells in nodules are well
differentiated and do not express atypia or increased mitotic level.
• cells that intersect slightly or ramify in the soft tissue, creating a
plexiform growth pattern.
• that the cells are usually plumper and the fascicles shorter than
those of fibromatosis.
Immunohistochemistry:
• muscle actin is positive in spindle cells.
Prognosis:
• 12-40% within 1-2 years of the original diagnosis and excision.
FETAL RHABDOMYOMA
Definition:
• muscle differentiation and predilection for the head and neck in
children.
Sites of involvement:
• mucosal sites of the head and neck
(most common postauricular site).
Clinical features:
• • • • soft tissue or mucosa of head and neck.
Gross:
• glistening cut surface.
Histopathology:
• 1) Myxoid classic type is composed of primitive oval or spindle-
shaped cells with indistinct cytoplasm, interspersed immature
skeletal muscle fibers reminiscent to fetal myotubes seen during
the seventh to tenth weeks of intrauterine life, in rich myxoid
stroma. The immature skeletal muscle cells have small uniform
nuclei with delicate chromatin and inconspicuous nucleoli with
bipolar or sometimes unipolar, eosinophilic cytoplasm. Cross
striations are rare and difficult to identified.
2) numerous differentiated muscle fibers, less conspicuous or absent
spindle-shaped mesenchymal cells, and little or no myxoid
stroma. The predominant cells are strap-shaped muscle cells with
abundand eosinophilic cytoplasm, centrally located vesicular
nuclei, and frequent cross-striations reminiscent of the cells seen
in adult rhabdomyomas; many of the cells contain glycogen and
are often vacuolated. Others have prominent ganglion-like
rhabdomyoblasts with large vesicular nucleai and prominent
nucleoli. In some cases there is mild cellular pleomorphism, but
marked cellular atypia is not seen. Transitional forms of myxoid
and intermediate types are not rare and in fact age and duration
may play a role.
Immunophenotype:
• muscle antigen.
Genetics:
• patients with nevoid basal cell carcinoma syndrome.
Prognosis:
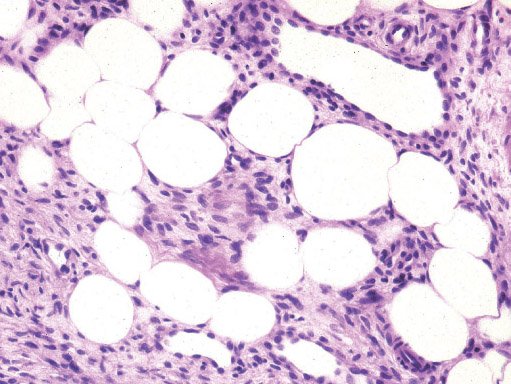
• LIPOBLASTOMA/LIPOBLASTOMATOSIS
Definition:
• tumor, resembling fetal adipose tissue.
Epidemiology:
• years of life.
• • Sites of involvement:
• mediastinum, retroperitoneum and head and neck areas was
described.
Clinical features:
• well circumscribed and confined to the subcutis in case of
lipoblastoma , infiltrating deeper muscle in case of
lipoblastomatosis.
Gross:
• gelatinous areas.
Histopathology:
• mature and immature adipocytes, the latter corresponding to
lipoblasts in various stages of development.
• and lesion may be dominated by lipoma-like component
(mostly in older patients).
• • • mimicking myxoid liposarcoma.
• hematopoiesis or cells resembling brown fat.
• picture.
Genetics:
• which has been found in majority of cases.
Prognosis:
• metastatsis does not occur.
• lipoblastomatosis.
The information provided on this site is designed to support, not replace, the relationship that exists between a patient/site visitor and his/her existing physician. Banners on the Home Page and at the top of other pages are advertisements. Our editorial content is free of any commercial influence. There are no medical or personal patient informations included. Disclaimer: this information is intended for pathologists, orthopedic surgeons and laboratory personnel, who understand that medical information is often imperfect, and must also be interpreted in the context of a patient's clinical data using reasonable medical judgment.
The site is supported by advertisement. I value our advertisers, who make this free website possible.
Pedorthpath.com is concerned with online privacy for all visitors and advertisers at our site. Any information sent to us by email or otherwise, except for information intended to be posted at the website, is treated as confidential, and will not be disclosed to any third party without the express permission of the visitor or advertiser.
Last modification date: 03/15/2010.
Site designed and maintained by Dariusz Borys, M.D
© Dariusz Borys 2009-2010. All Rights Reserved.
This page was last updated: May 15, 2016
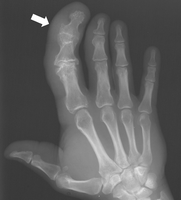
LIPOMATOSIS OF NERVE (FIBROLIPOMATOUS HAMARTOMA)
Definition:
• epineurium by adipose and fibrous tissue.
• enlargement of involved nerve.
Epidemiology:
• • y/o. In some cases it is associated with macrodactyly of the
digits innervated by the affected nerve.
• macrodactyly, whereas males are more commonly affected
when macrodactyly is absent.
Sites of involvement:
• affected followed by the ulnar nerve.
Clinical features:
• that may be asymptomatic or associated with motor or
sensory deficits.
• enlargementf of affected finger(s) with enlargement of the
involved bones.
Imaging:
• Gross:
• fibrofatty tissue, which is generally confined within the
epineural sheath.
Histopathology:
• are infiltrated by mature adipose tissue which dissects and
separate nerve bundles.
• • septation, microfascicle formation and pseudoonion bulb
formation mimicking an intraneural perineurioma.
Prognosis:
• • involved nerve.
• neurological symptoms.